

La alfa-manosidosis es una enfermedad huérfana en niños. Conoce sus principales síntomas y la importancia de un diagnóstico medico oportuno
¿Qué es la alfa-manosidosis?1
La alfa-manosidosis es una enfermedad huérfana (infrecuente), de origen genético, clasificada en el grupo de patologías de depósito lisosomal. Estas enfermedades ocurren cuando el cuerpo no produce una enzima encargada de transformar algunos compuestos específicos de las células (en este caso llamados oligosacáridos). Esto ocasiona que se acumulen, causen daño en tejidos y órganos del cuerpo.
Esta enfermedad se puede dividir en 3 formas, dependiendo de la severidad de los síntomas:
- Tipo 1: Forma leve de la enfermedad que se caracteriza porque aparece luego de los 10 años de vida y avanza progresivamente.
- Tipo 2: La enfermedad también progresa lentamente, pero los síntomas empiezan a aparecer antes de los 10 años de vida.
- Tipo 3: Forma más severa, aparece tempranamente, habitualmente antes de los 5 años de edad y puede resultar en la pérdida del embarazo o la muerte del recién nacido.
¿Qué tan común es la alfa-manosidosis?2
La alfa-manosidosis es infrecuente, se estima que puede afectar a 1 de cada 1.000.000 de personas en el mundo.
¿Cuáles son los síntomas de la alfa-manosidosis?
Los síntomas son variables pero algunos son más frecuentes:3
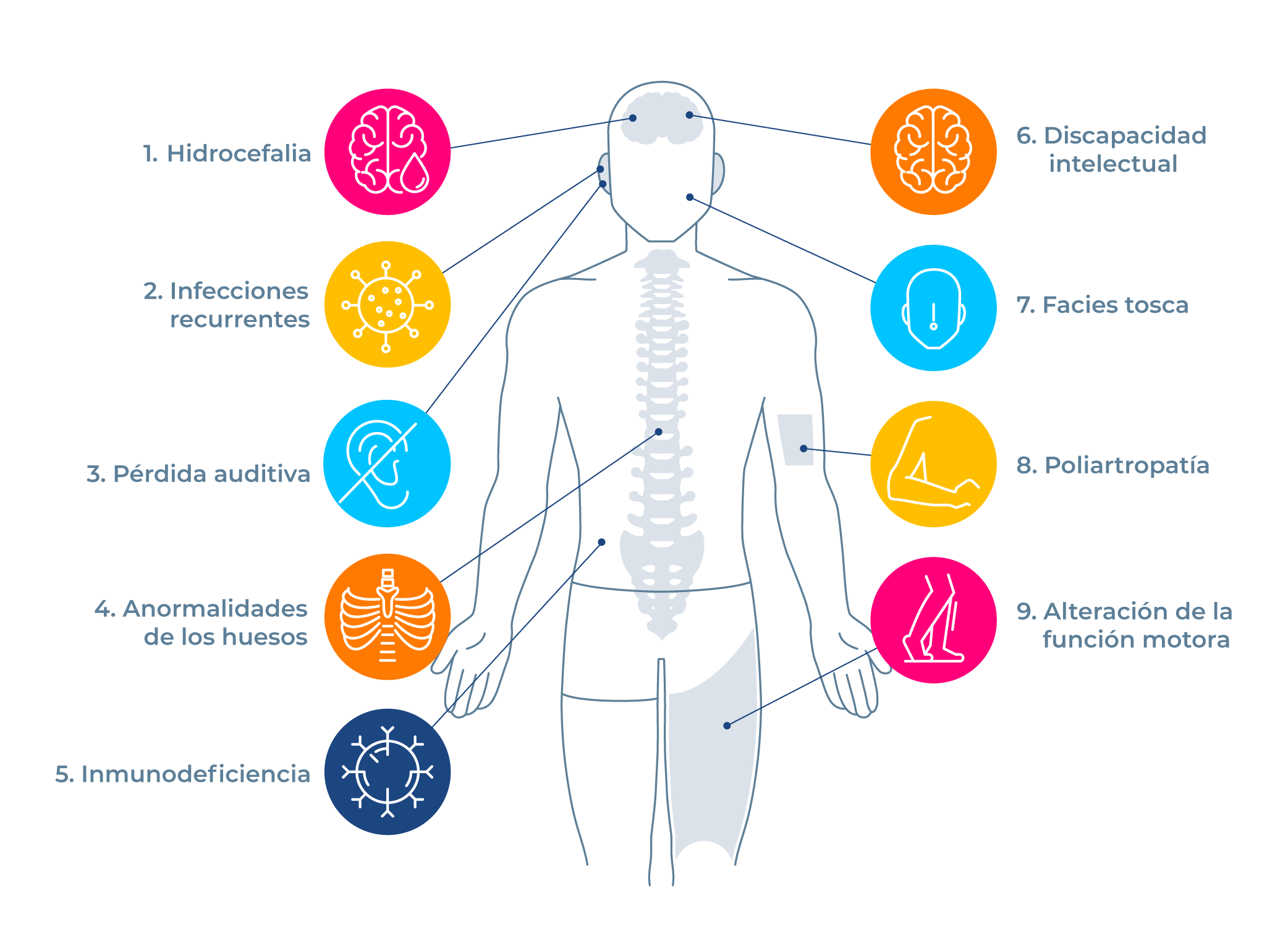
Discapacidad intelectual: La inteligencia y la capacidad para desenvolverse en las actividades diarias están por debajo del nivel promedio, incluyendo las habilidades prácticas, sociales y conceptuales.
Hidrocefalia: El líquido que se encuentra en el cerebro (líquido cefalorraquídeo) se acumula de forma excesiva y ejerce una presión dañina para el cerebro.4
Infecciones recurrentes: Las personas presentan infecciones a repetición, estas pueden ser urinarias, pulmonares y de oído entre las más comunes.
Pérdida auditiva: Puede existir una disminución de la audición de forma parcial o completa.
Poliartropatía: alteración en 2 o más articulaciones del cuerpo, puede presentarse dolor, inflamación y/o deformidad.
Anormalidades de los huesos: Los huesos de la columna vertebral, las costillas, los brazos, las manos, las piernas y la pelvis se desarrollan de forma irregular, causando limitaciones, dolor o deformidad.
Alteración de la función motora: Se presenta por daños en el cerebro, los nervios o los músculos, puede haber debilidad muscular y alteraciones en la coordinación que afectan la marcha, las extremidades y el lenguaje. Las personas pueden parecer torpes al caminar.
Inmunodeficiencia: El sistema encargado de la defensa del cuerpo a las infecciones (sistema inmunológico) no funciona correctamente, siendo los pacientes más vulnerables a las infecciones por los virus, las bacterias o los hongos. Se presentan infecciones a repetición.
Facies tosca: frente prominente, párpados hinchados, fosas nasales anchas, implantación baja de las orejas, labios gruesos, lengua grande, puente nasal ancho, cejas pobladas y arqueadas.

Estos síntomas pueden cambiar a medida que los pacientes envejecen. En la infancia, es común que tengan pérdida auditiva, retraso en el neurodesarrollo (que tengan dificultades para empezar a hablar y/o caminar, que se demoren o no logren hacer las cosas que ya hacen todos los niños de su edad), hidrocefalia en el primer año de vida e infecciones recurrentes (especialmente urinarias, pulmonares y del oído).
En la segunda y tercera década de la vida, pueden esta afectados además por debilidad muscular o ataxia (problemas con el equilibrio y la coordinación). Los pacientes que sobrepasan los 30 años de vida son generalmente muy dependientes de otras personas en la vida cotidiana. Es difícil para ellos realizar actividades por si solos debido a diferentes discapacidades1,2
¿Cómo es vivir con alfa-manosidosis?
La alfa-manosidosis impacta de forma significativa la calidad de vida de los pacientes y sus familias, requiriendo un alto nivel de cuidado, tiempo y recursos. Por los síntomas asociados a la enfermedad, los pacientes pueden requerir de múltiples intervenciones a lo largo de su vida: implantación de dispositivos auditivos, cirugías, uso de sillas de ruedas, constante rehabilitación física. Además, requieren que los espacios de sus hogares deban adaptarse especialmente para facilitar la movilidad. Terapias de lenguaje y físicas son requeridas.5,6,7,8
¿A quién deben consultar las personas que sospechen tener alfa-manosidosis?
Si una persona está afectada por síntomas que puedan presentarse en la alfa-manosidosis, lo más recomendable es que sea valorado por médicos especializados en pediatría, neurología pediátrica y/o genética. Estos especialistas realizarán una valoración detallada que puede incluir un examen enzimático o genético que permita el diagnóstico definitivo de la enfermedad.
En la actualidad existen tratamientos para esta enfermedad. La respuesta depende, entre otros, de la oportunidad temprana del diagnóstico y el acceso al tratamiento.
En la actualidad existen 2 alternativas principales:
- Trasplante de medula ósea:Es un procedimiento invasivo que consiste en trasplantar células madre sanas en la médula ósea del paciente con la capacidad de diferenciarse en diferentes tipos de células y producir la enzima que está deficiente en el cuerpo. Este tratamiento generalmente es definitivo
- Terapia de reemplazo enzimático: Este tipo de terapia reemplaza por vía intravenosa la enzima que se encuentra deficiente en el cuerpo. Este tratamiento debe ser administrado de por vida o antes de que se encuentre un donante para efectuar el trasplante descrito anteriormente.
Para todas estas terapias existen indicaciones, contraindicaciones y complicaciones. El médico es quien determina la indicación para realizar un tratamiento específico.
Si estás en Colombia, Argentina, Chile o Perú y presentas dudas sobre la alfa-manosidosis contáctate a [email protected] para recibir más información y orientación.
Referencia:
- Malm D et al. Orphanet J Rare Dis 2008;3:21.
- Borgwardt L et al. Pediatr Endocrinol Rev 2014;12(Suppl 1):185–191.
- Beck M et al. Orphanet J Rare Dis 2013;8:88.
- MedlinePlus. Alpha-mannosidosis. 2020. Available from: https://medlineplus.gov/genetics/condition/alpha-mannosidosis/
- Adam J et al. Mol Genet Metab Rep 2019;20:100480. doi: 10.1016/j.ymgmr.2019.100480. eCollection 2019 Sep.
- National Institute for Health and Care Excellence. Evaluation consultation document Velmanase alfa for treating alpha-mannosidosis, May 2018. Available from: https://www.nice.org.uk/guidance/GID-HST10010/documents/evaluation-consultation-document. Accessed October 2020.
- Guffon N et al. Mol Genet Metab 2019;126(4):470–474.
- National Institute for Health and Care Excellence. Highly Specialised Technology Evaluation. Velmanase alfa for treating alpha-mannosidosis [ID800]. Evaluation Report, February 2020. Available from: https://www.nice.org.uk/guidance/gid-hst10010/documents/committee-papers. Accessed October 2020.
- MedLink Neurology (2022)- Mannosidosis. [Image] Recuperada de: https://www.medlink.com/articles/mannosidosis
AV-VAR-VAR-N1348-2022-Vig.NOV2024-VAR


